FALLA OVARICA PREMATURA Y AUTOINMUNIDAD
Autor: Ana Carolina Robín
INTRODUCCION
La falla ovárica prematura (FOP) es un desorden heterogéneo con una patogénesis multicausal (4)(Tabla 1) cuya base es la desaparición o la disfunción de folículos en el ovario en mujeres menores de 40 años (1)(2)(8)(11)(22)(39) . Es una entidad caracterizada por:
- amenorrea (al menos de 4 meses) (1)
- deficiencia estrogénica y
- elevados niveles de gonadotrofinas (FSH mayor a 40 mUI/ml en dos o más oportunidades) (3).
La menopausia, cese de la menstruación sobre la base de la depleción folicular, es un evento fisiológico, que ocurre a un promedio de edad de 50 años (4) y resulta una condición irreversible, mientras la falla ovárica prematura está caracterizada por una función ovárica intermitente (13)(34).
La elección de los 40 años de edad para separar menopausia prematura de menopausia fisiológica es arbitraria. Si se definiera como anormal aquellos valores por encima o por debajo de dos desvíos estándares de la edad media de la menopausia, la edad de 43 años sería la más apropiada como límite inferior para el cese natural de la menstruación (4).
El número de folículos primordiales disminuye con la edad y el último en desaparecer en el ovario es el responsable del cese de la función ovárica en la menopausia(38). Sin embargo han sido encontrados, por microscopía electrónica, oocitos normales en ovarios de mujeres postmenopaúsicas, indicando que la desaparición de los mismos puede no ser el único factor que juegue un rol en el desarrollo de este estado(4). Algunos autores postulan que estos folículos remanentes en mujeres post menopaúsicas son menos sensibles a los altos niveles de gonadotrofinas (40) .
|
Tabla 1 Causas de falla ovárica prematura (1)(2) Depleción del folículo ovárico 1. Deficiente número inicial de folículos
2. Atresia acelerada
Disfunción del folículo 1-Deficiencias enzimáticas
2-Autoinmunidad 3-Ooforitis linfocítica
4-Defectos en la señal
5-Iatrogénica (quimioterapia-radiación) 6-Idiopática Síndromes raros asociados a FOP 1-Blefarofimosis, ptosis y epicantus inversus, síndrome tipo 1 2-Síndrome de Perrault
|
 -hidroxilasa
-hidroxilasaEn la mayoría de los pacientes la falla ovárica prematura es idiopática. Sin embargo en unos pocos pacientes puede reconocerse un proceso de enfermedad específico. Debido a que la etiología puede alterar el plan de tratamiento es recomendable determinar la causa de la falla ovárica prematura. (1)
INCIDENCIA
La incidencia de FOP es de 1% en mujeres menores de 40 años y de 0.1% en mujeres menores de 30 años. (20)
En mujeres con amenorrea primaria la prevalencia de FOP es de 10-28 %; en aquellas con amenorrea secundaria entre un 4-18 % (2)(19) . No es un desorden poco común teniendo en cuenta la incidencia y el número de mujeres que no perciben la falta del sangrado menstrual como un problema médico. (2)
PRESENTACIÓN CLINICA
El diagnóstico de FOP se realiza en base a los signos clínicos y a los niveles de FSH superiores a 40 UI/L, con respecto al 2° IRP ( Preparación Internacional de Referencia, Gonadotrofina Menopaúsica Humana; IRP-hMG) en dos o tres oportunidades separadas por el lapso de un mes (2) ( 4).
El cese de la función ovárica puede ocurrir a cualquier edad, originando amenorrea primaria si es antes del desarrollo puberal o amenorrea secundaria luego de la menarca(3). La falla para completar el desarrollo de caracteres sexuales secundarios y las anormalidades cromosómicas son más frecuentes en pacientes con amenorrea primaria, quienes pueden tener deleciones de todo o una parte del cormosoma X, mientras que aquellas con amenorrea secundaria comúnmente tiene un cromosoma X adicional.
La edad de presentación depende en muchos casos del momento y la rapidez de la atresia folicular. De un total de 115 pacientes evaluadas al momento de la presentación, aquellas con amenorrea primaria tenían una edad media de 22,7 años, y aquellas con amenorrea secundaria tenían 27,9 años. El momento preciso de su comienzo es imposible de determinar.(3)
La FOP no siempre es permanente, pudiendo existir formas subclínicas o latentes de este desorden(6). La enfermedad puede tener un curso fluctuante con niveles de gonadotrofinas elevados que luego retornan a la normalidad, con recuperación de la función ovárica, ovulación y aún en algunos casos concepción (13)(19)(30) .
No hay una historia menstrual característica que preceda a la falla ovárica prematura. Aproximadamente el 50% de las pacientes tienen oligomenorrea o un sangrado uterino disfuncional (pródromo de FOP), el 25% tiene amenorrea aguda, algunas post parto y otras luego de discontinuar la terapia anticonceptiva. (4)(28)(30)
Los síntomas en casos de amenorrea secundaria pueden incluir: síntomas vasomotores , sudor nocturno, cambio de humor, atrofia de la vagina y del tracto urogenital, conduciendo a vaginitis, dispaurenia y cistitis, osteoporosis e infertilidad debido al estado hipoestrogénico (1)(2)(4)(11)(19)(22,. Mientras que la amenorrea primaria no está asociada con síntomas de deficiencia estrógenica
FORMAS HISTOPATOLÓGICAS
Kinch fue el primero en identificar dos tipos histopatológicos de falla ovárica prematura:
1. la forma afolicular y
2. la forma folicular.
En la forma afolicular hay una depleción total de los folículos con una pérdida definitiva de la función ovárica. Es debida principalmente a disgenesias gonadales, gonadoblastoma mixto, y hermafroditismo(22). Las anormalidades genéticas y cromosómicas son las causas fundamentales de desaparición o mal desarrollo de las células germinales, mientras una acelerada pérdida de los oocitos es la causa de FOP en individuos con cariotipo 47XXX, 45XO,o mosaicismo 45XO. Estudios en individuos 45X0 han demostrado que son necesarios dos cromosomas X intactos para el mantenimiento más que para la diferenciación de los oocitos. Los ovarios de fetos 45X0, por ejemplo, contienen un número normal de folículos que degeneran en forma rápida al nacer.
En la forma folicular las estructuras foliculares están preservadas y es posible el retorno de la función ovárica en forma espontánea o inducida.
La forma folicular puede ser subdividida en :
- · ooforitis (inflamación del folículo)
- · ovarios con pocos folículos presentes
- · ovarios con numerosos folículos presentes ( síndrome de ovario resistente).
Síndrome de ovario resistente (SOR)
En 1969 Moraes-Ruehsen y Jones (43) describieron tres pacientes con falla ovárica prematura, pero con aparato folicular normal, resistentes a la terapia inductora de la ovulación (citrato de clomifeno, gonadotrofinas menopaúsica humana). Esta manifestación de resistencia ovárica fue llamada síndrome de Savage, en honor a la primera paciente estudiada.
Los pacientes con síndrome de ovario resistente tienen amenorrea primaria o secundaria, usualmente con desarrollo normal de caracteres sexuales secundarios, y elevados niveles de gonadotrofinas en presencia de numerosos folículos primordiales morfológicamente normales, bajos niveles séricos de estradiol y carencia de respuesta a gonadotrofinas exógenas (15)(28)(30)(41)(42)(43)(48)(51). En contraste a la situación en mujeres postmenopaúsicas la biopsia de ovario demuestra la presencia de múltiples folículos inmaduros. La patofisiología del síndrome no está bien entendida, aunque muchas hipótesis han sido postuladas. Desde la carencia de receptores a gonadotrofinas, disturbios en las vías post receptor, gonadotrofinas con inadecuada bioactividad, factores séricos que modulen la acción de FSH y factores inmunes como anticuerpos a receptores de gonadotrofinas y patología tímica.
Aunque la clasificación histólogica sugiere una marcada división entre las formas foliculares y afoliculares de falla ovárica prematura, hay evidencia de algunos casos que se iniciaron como folicular y progresaron hacia un estadío afolicular (4).Trabajos recientes sugieren que la autoinmunidad ovárica es una posible causa de ambas formas (folicular y afolicular) de FOP.
AUTOINMUNIDAD Y FALLA OVÁRICA PREMATURA
La más importante función del sistema inmune es reconocer
lo propio de lo no propio. Se denomina autoinmunidad a la respuesta inmune
contra componentes propios, y es debido a la presencia de linfocitos T
o B autorreactivos. Para entender la autoinmunidad es importante primero
entender la tolerancia. La tolerancia puede ser definida como un estado
de falta de respuesta inmunológica inducida por un antígeno(11).
Puede ser transitoria o permanente. En condiciones normales, los mecanismos
de tolerancia a lo propio protegen al individuo de estas células
potencialmente autorreactivas. Las enfermedades autoinmunes se caracterizan
por la presencia de autoanticuerpos y/o linfocitos T sensibilizados contra
diferentes macromoléculas del organismo. La respuesta inmune a
antígenos propios se puede observar en individuos normales, pero
la potencialidad para desarrollar autoinmunidad está controlada
por diferentes mecanismos: linfocitos T supresores, interacciones idiotipo-antiidiotipo
, bloqueo de receptores celulares por antígenos circulantes,etc.
En la enfermedad autoinmune la presencia de autoanticuerpos o linfocitos
T sensibilizados producen lesión tisular y disfunción de
los órganos blanco , mecanismos totalmente descontrolados y generalmente
irreversibles.
Hay evidencia acumulada indicando que algunos casos de FOP son debidos
a la falta de reconocimiento, por el sistema inmune, de lo propio en el
ovario. (Tabla 2). Algunos investigadores estiman indicar que a lo sumo
el 15-30% de los casos de FOP son debido a enfermedad autoinmune (11)
.
Tabla 2
Evidencia de autoinmunidad en la etiología de FOP
· Asociación con otras enfermedades autoinmunes
· Presencia de anticuerpos circulantes
1-Anticuerpos antiovarios
2-Anticuerpos contra el receptor de gonadotrofinas
3-Anticuerpos anticélulas esteroideas
4-Anticuerpos contra la zona pelúcida
5-Otros anticuerpos órgano y no-órgano específico
· Evidencia histológica de ooforitis
· Asociación con etiología infecciosa
· Alterada inmunidad mediada por células
· Recuperación de la función ovárica luego de terapia inmunosupresiva
· Falla ovárica inducida por timectomía en modelos neonatales murinos
Tolerancia a lo propio
En individuos sanos la reactividad hacia lo propio es actualmente considerado un evento normal que es controlado por múltiples mecanismos de “down-regulation”. El mal funcionamiento de estos mecanismos puede resultar en una, no deseada, reacción inmune excesiva hacia lo propio por ej: enfermedad autoinmune. Los mecanismos de control son los siguientes:
1. Deleción clonal en el timo
2. Anergia clonal
3. Inmunosupresión activa (11)
Deleción => anergia clonal => inmunosupresión activa
Deleción clonal:
Las células T maduran en el timo de protimocitos a células T maduras. Debido a que el reordenamiento del receptor T es aleatorio, se generan en este proceso células T autorreactivas. Sin embargo, la amplia mayoría son delecionadas en el timo durante el proceso de maduración. La deleción ocurre en gran número en las uniones córtico-medulares del timo, donde se expresan antígenos propios y este proceso depende del reconocimiento de péptidos antigénicos propios por células T no maduras totalmente, lo cual conduce a apoptosis. Un mecanismo similar puede existir para las células B en la médula ósea. Aunque no está tan estudiado.
Si la deleción clonal es incompleta, células T y B con especificidad para péptidos autoantigénicos y autoantígenos pueden ser fácilmente encontrados en la circulación. Esto es explicado parcialmente por el hecho de que no todos los antígenos propios son expresados en la médula ósea o en el timo, particularmente se aplica a los epitopes crípticos.
Anergia clonal
Cuando células T o B autorreactivas han escapado a la deleción clonal, un segundo mecanismo de control anergia clonal puede entrar en operación. Este proceso tiene lugar en la periferia. La inducción de un estado anérgico inmunológicamente de la célula T se postula es debido a la carencia de provisión de suficientes segundas señales por la célula presentadora de antígenos. Así, cuando el antígeno es presentado a la célula T por una célula presentadora de antígenos no profesional, como una célula epitelial CMH clase II positiva, la anergia clonal puede ocurrir. En la enfermedad autoinmune órgano específica hay una expresión aberrante de complejos moleculares de histocompatibilidad clase II sobre la célula epitelial del tejido afectado. Esto fue interpretado como un ímpetu de incrementar la reactividad hacia lo propio, sin embargo esto puede ser considerado como una señal para inducir anergia clonal.
Inmunosupresión activa:
Cuando la deleción y anergia clonal han fallado aún puede entrar en operación otro mecanismo de “down regulation” llamado inmunosupresión activa, ejercida por células inmunes llamadas supresoras. Estas células involucradas pueden ser o no antígeno específicas o/y operar de una forma del tipo idiotipo-antiidiotipo. Las células inmune supresoras no sólo incluyen células T CD4+ y CD8+ sino también macrófagos supresores. El mecanismo no es claro. Recientes evidencias sugieren que en cada individuo hay un balance entre las células inmunes supresoras y los efectores autorreactivos, inclinándose a favor de estos últimos en el estado de enfermedad autoinmune.
Balance entre TH-1( T helper 1) y TH-2( T helper 2)
Esta teoría aborda el problema como una falla de la glándula
blanco causada por una vía mediada por Th1 en la cual la célula
endócrina es destruida por macrófagos scavenger activados
por interferón gamma (IFN-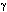 ). La teoría
recientemente desarrollada enfatiza la relación recíproca
entre las vías Th1 y Th2 y sugiere que si la vía Th1 es
desviada hacia la vía Th2 , la reactividad autoinmune mediada por
Th1 es “desalentada” (4).
). La teoría
recientemente desarrollada enfatiza la relación recíproca
entre las vías Th1 y Th2 y sugiere que si la vía Th1 es
desviada hacia la vía Th2 , la reactividad autoinmune mediada por
Th1 es “desalentada” (4).
ENFERMADAD ENDÓCRINA AUTOINMUNE
Se carece de modelos animales para la ooforitis y adrenalitis autoinmune espontánea. Sólo manipulaciones de ratones normales ( inmunizados con extractos ováricos o adrenales crudos, y timectomía más tratamiento con ciclosporina A ) pueden conducir a adrenalitis y ooforitis autoinmune . Se requiere precaución cuando se extrapolan datos obtenidos en modelos animales al ser humano.(1)(2)
El modelo animal de insulinitis (DMID) y tiroiditis (falla tiroidea) indica que la patogénesis de la falla autoinmune de una glándula endócrina es un proceso de múltiples pasos, que requiere de anormalidades genéticas y ambientales juntas antes del desarrollo de la insulinitis y tiroiditis.
Se pueden distinguir las siguientes fases en el desarrollo de la enfermedad:
1. Fase inicial: acumulación de células presentadoras de antígeno y células accesorias en el tejido endócrino
2. Fase más tardía: aparente descontrolada producción de células T CD8+ y CD4+ (expansión clonal) y de autoanticuerpos de clase IgG (inmunoglobulina G) en el drenaje del nódulo linfático.
3. Última fase: donde el tejido endócrino blanco comienza a ser suceptible al ataque por las células T autorreactivas y los autoanticuerpos , finalmente resulta en la destrucción del tejido glandular.
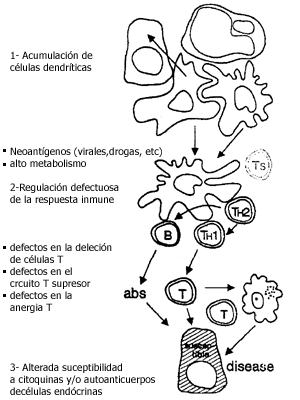
1- Acumulación de células dendrítícas:
· Neoantígenos (virales,drogas, etc)
· Alto metabolismo2-Regulación defectuosa de la respuesta inmune:
· defectos en la deleción de células T
· defectos en el circuito T supresor
· defectos en la anergia T3- Alterada suceptibilidad a citoquinas y/o autoanticuerpos de células endócrinas
Se han descripto como posibles causas de la atracción de células dendríticas al tejido endócrino : antígenos extraños de origen viral o bacteriano, antígenos propios alterados por toxinas y drogas o una excesiva actividad metabólica del tejido involucrado, demostrando la heterogeneidad de factores causales a nivel de la iniciación de la enfermedad endócrina autoinmune.
La anormalidad inmune sistémica está en parte asociada con la presencia de determinados haplotipos del Complejo Mayor de Histocompatibilidad (CMH) clase I y clase II y aparentemente conduce a anormalidades en la estimulación y diferenciación de células involucradas en la inducción de tolerancia, así como células presentadora de antígenos, macrófagos, y/o células T. Hay numerosos reportes sobre defectos funcionales y numéricos en las células T supresoras de pacientes con DMID y tiroiditis (4).
Este déficit en las células inmunorreguladoras no sólo puede ser heredado genéticamente sino también puede ser adquirido por infecciones virales (fetales). Está probado en animales , pero aún en humanos no son concluyentes los experimentos para demostrar en qué medida están involucrados los virus en la enfermedad endócrina autoinmune.
Un prerrequisito para el desarrollo de la enfermedad autoinmune es la susceptibilidad de la glándula blanco. Estudios poblacionales, en familias y gemelos han mostrado claramente que los factores genéticos ejercen una influencia significativa sobre la predisposición para una enfermedad endócrina autoinmune. Es claro también que factores ambientales (dieta, infecciones, etc) contribuyen a la expresión de la enfermedad, de acuerdo a estudios en gemelos monocigotas.
Asociación con otras enfermedades autoinmunes
Ya desde 1950 se ha notado que el comienzo de la FOP precede, sucede o se desarrolla junto con el comienzo de otra enfermedad endócrina o no endócrina que involucre la autoinmunidad (Tabla 3)(2)(3)(4).
Tabla 3
Alopecía
Anemia hemolítica adquirida y perniciosa
Asma
Hepatitis crónica activa
Enfermedad de Crohn
Diabetes mellitus
Glomerulonefritis
Hipoadrenalismo (Enfermedad de Addison)
Hipoparatiroidismo
Hipofisitis
Púrpura trombocitopénica idiopática
Artritis reumatoidea juvenil
Queratoconjuntivitis y Síndrome de Sjögren
Síindrome de malabsorción
Mistenia gravis
Vitiligo
Un total de 119 casos en los cuales la FOP está asociada con alguna enfermedad autoinmune son mostrados en la tabla 4 (8).
Tabla 4 Números de casos reportados con FOP y enfermedad autoinmune asociada(8)
| Estudio | FOP * | Enf.autoinmune | Tiroides | Adrenal | SPA tipo I | SPA tipo II |
| Total casos | 380 | 119 | 26 | 8 | 20 | 19 |
| Estudio | DM | Endocrinopatía múltiple | M.gravis | A. Perniciosa | No especificado | Otro |
| Total casos | 3 | 6 | 9 | 2 | 15 | 11 |
*Excluídos los casos con cariotipo anormal
SPA tipo I: Síndrome poliglandular tipo I
SPA tipo II: Síndrome poliglandular tipo II
DM: diabetes mellitus, M.gravis: miastenia gravis, A.Perniciosa: anemia
perniciosa
La frecuencia de asociación varía desde 0 a 57% con una término medio de 17,5%. Esta variación puede ser explicada parcialmente por una diferente evaluación del paciente. Un mayor porcentaje de asociación se obtiene de reportes que excluyen todos los pacientes con cariotipo anormal o incluyen pacientes con enfermedades subclínicas (8) .
En los reportes más tempranos uno de los primeros indicios a favor de la causa autoinmune en la falla de la función ovárica es la observación de que la misma puede preceder en 8 a 14 años el comienzo de la enfermedad de Addison (2). Se estima que el 10% de los pacientes desarrollan FOP. Desde entonces muchos investigadores han buscado la asociación con falla ovárica y desórdenes autoinmunes.
La enfermedad de Addison es un desorden poco común (afecta 10-20 personas por millon de habitantes) causado por una insuficiencia en la producción de hormonas adrenales. En los países desarrollados la mayoría de los pacientes con enfermedad de Addison son ahora considerados como autoinmune, en contraste a los países en desarrollo donde se acusa principalmente a la tuberculosis. La enfermedad de Addison autoinmune rara vez se desarrolla en forma aislada y generalmente están afectadas glándulas endócrinas y otros órganos, conduciendo al Síndrome Poliglandular Autoinmune (SPA)(tabla 5). (Cuando la disfunción de dos o más glándulas endócrinas ocurre en asociación con anticuerpos órgano específicos dirigidos contra la glándula se aplica el término síndrome poliglandular autoinmune).
Se distinguen clínicamente dos formas principales, el SPA tipo I, que afecta exclusivamente a chicos y adultos jóvenes, parece ser secundario a una disfunción tímica. Está caracterizado por la asociación de dos de las siguientes tres condiciones: candidiasis mucocutánea, hipoparatiroidismo y enfermedad de Addison y menos frecuentemente diabetes mellitus insulino dependiente (tabla 6). Se hereda de forma autosómica recesiva. Las primeras manifestaciones de este síndrome se desarrollan antes de los quince años. La edad promedio de aparición de la candidiasis y/o el hipoparatiroidismo son los cinco años y precede típicamente a la aparición de insuficiencia adrenal, la cual generalmente ocurre a un promedio de edad de 13 años (11). La falla ovárica es usualmente parte del síndrome (aproximadamente entre el 45- 60% de los casos) (2)(4)(11). Se manifiesta generalmente como amenorrea primaria. La amenorrea secundaria ocurre antes del diagnóstico de enfermedad de Addison y luego del comienzo del hipoparatiroidismo. La prevalencia incrementada, cosanguineidad y el análisis frecuente en miembros de una familia afectada sugiere un modo autosómico recesivo de herencia (11). Las mujeres están frecuentemente más afectadas que los hombres. Se ha postulado que el defecto genético en este Síndrome este relacionado a una disminuída regulación de las células T supresoras (11).
Tabla 5 Principales manifestaciones de los Síndromes poliglandulares autoinmunes asociados con enfemedad de Addison (11)
Síndromes asociados con la enfermedad de Addison
SPA tipo I
Hipoparatiroidismo
Candidiasis mucocutánea crónica
Insuficiencia adrenal
SPA tipo II
Insuficiencia adrenal
Enfermedad tiroidea autoinmune
Diabetes mellitus insulino dependiente
Tabla 6 Manifestaciones endócrinas asociadas al SPA tipo I
Manifestaciones endócrinas del síndrome poliglandular
autoinmune tipo I
Prevalencia en Desórdenes endócrinos
Hipoparatiroidismo 89%
Candidiasis mucocutánea crónica 75%
Insuficiencia adrenal 60%
Falla gonadal 45%
Enfermedad tiroidea autoinmune 12%
Diabetes mellitus insulino dependiente 1%
Hipopituitarismo-diabetes Insípida <1%
Diabetes Insípida <1%
El Síndrome poliglandular autoinmune tipo II está caracterizado por insuficiencia adrenal en asociación con hipotiroisimo y/o diabetes mellitus insulino dependiente (tabla 7) (11). Ocurre en 1 de 20000 individuos, siendo más frecuente que el SPA tipo I. Este último principalmente se manifiesta en la cuarta década de la vida y preponderantemente en mujeres (4). En este Síndrome y de acuerdo al autor, entre un 25% a un 50% de las mujeres tienen amenorrea y un 10% presentan la forma clásica de FOP(4)(11). Con respecto a la FOP sólo el 2-10% está asociado con la enfermedad de Addison y/o autoinmunidad adrenal(4)(8)(29).Sin embargo, estudios más recientes sugieren que la enfermedad tiroidea es la endocrinopatía autoinmune predominante asociada con FOP.(8)(9) Esto puede reflejar cambios en el patrón de asociación, mejoras en las técnicas de diagnóstico de la enfermedad tiroidea subclínica o un incrementado conocimiento de la necesidad de screening de enfermedades autoinmunes asociadas(8).
El tipo de enfermedad endócrina autoinmune asociada con FOP ha sido especificado en 8 series de pacientes desde 1980. De las 263 pacientes estudiadas, el 9% tenía enfermedad tiroidea, el 35 poliendocrinopatía, el 2% enfermedad de Addison, el 1% artritis reumatoidea y menos del 1% lupus eritematoso sistémico, asma, glomerulonefritis, vitiligo, miastenia gravis, diabetes mellitus o enfermedad de Crohn(2). La incidencia combinada de enfermedad autoinmune fue del 20%.
Tabla 7 Manifestaciones endócrinas asociadas al SPA tipo II
Manifestaciones endócrinas en el síndrome ploglandular
autoinmune tipo II
Prevalencia en Desórden endócrino
Insuficiencia adrenal 100%
Enfermedad tiroidea autoinmune 70%
Diabetes mellitus insulino-dependiente 50%
Falla gonadal 25-50%
Diabetes Insípida 20-50%
La FOP está asociada a autoinmunidad no-órgano específica sólo raramente, con cinco casos reportados en la literatura especializada. Tres casos diagnosticados con lupus eritematoso sistémico y dos con artritis reumatoidea (3).
MARCADORES INMUNOGENÉTICOS
Al presente no hay evidencia convincente de una relación entre la predisposición a la enfermedad endócrina autoinmune y un particular haplotipo o polimorfismo del receptor T, alotipos e idiotipos de inmunoglobulinas o citoquinas. Está generalmente aceptado que la enfermedad autoinmune no es una enfermedad genética por sí misma, pero ocurre en individuos suceptibles genéticamente. La observación que individuos con una enfermedad autoinmune comúnmente desarrollan una o más enfermedades autoinmunes adicionales sugiere una suceptibilidad genética a la autoinmunidad. Es conocida la asociación entre endocrinopatía autoinmune y antígenos HLA(35)(36). Hay fuertes evidencias que el antígeno HLA-DR y en particular el HLA-DR3 y DR-4 determinan una suceptibilidad individual a la enfermedad (2). Sin embargo Anasti y col. (35) no han podido demostrar el significativo incremento en la frecuencia de HLA-DR3 en el CMH en un estudio llevado a cabo en 102 pacientes con FOP. En contraposición Walfish y col.. (11) en otro trabajo sugieren que el 50% de las mujeres con FOP fueron HLA-DR3+ comparado con sólo el 23% de los controles.
La enfermedad autoinmune adrenal, tiroiditis y la insulinitis están predominantemente asociadas con los haplotipos HLA-DR3, DR4 y DR5. En un pequeño estudio de pacientes con SPA tipo I y tipo II el HLA-A9 fue más frecuente en aquellos con SPA tipo II con falla ovárica(25) . Estudios sobre varias familias con SPA tipo II y enfermedad de Addison idiopática han indicado una asociación con HLA-B8 y HLA-DR3 (25) . Mientras el HLA A28 se encontró en algunos en pacientes con SPA tipo I.
El CMH puede afectar la predisposición a la enfermedad endócrina autoinmune por muchos mecanismos. Los péptidos de autoantígenos glandulares pueden combinarse más fácilmente con estas moléculas del CMH en particular que con otras (8).
Sin reparar en los mecanismos, es evidente que el haplotipo del CMH ²per se² es insuficiente para desarrollar una enfermedad endócrina autoinmune, como es demostrado por el hecho que haplotipos HLA-DR asociados a autoinmunidad son encontrados también en individuos perfectamente normales(4).
Marcadores inmunogenéticos además del HLA podrían también contribuir a la susceptibilidad a la enfermedad autoinmune o afectar la expresión de la enfermedad en algunos individuos. Por ejemplo el efecto de inmunoglobulinas dependientes de HLA ha sido demostrado en familias con diabetes mellitus insulino dependiente. Efectos interactivos entre HLA y otros marcadores inmunogenéticos no han sido investigados en pacientes con FOP(8).
Presencia de anticuerpos circulantes
La presencia de anticuerpos a proteínas celulares específicas de órganos afectados en el suero de un paciente fortalece el diagnóstico de enfermedad autoinmune (7)(8), aunque la mera presencia de anticuerpos no indica citotoxicidad (8). Los autoanticuerpos pueden ser el agente patogénico de una enfermedad Ej: anemia hemolítica autoinmune, o pueden aumentar como consecuencia de otro proceso por ej. anticuerpos que reaccionan contra el músculo cardíaco luego del daño de este órgano o pueden meramente marcar la presencia de un agente etiológico mientras no causan daño por sí mismos (2)(11) .La detección de anticuerpos circulantes anti-ovario en pacientes con FOP ha sido contradictoria en diferentes publicaciones. La interpretación de su papel es problemático. La heterogeneidad de los métodos de investigación contribuye a la confusión de su importancia en la falla ovárica. Aunque pacientes con FOP tenían incrementada incidencia de anticuerpos a antígenos ováricos algunos investigadores encontraron que los mismos sueros contenían inmunoglobulina G que exhibía significativa unión a antígenos no ováricos (10).
Las técnicas comúnmente empleadas para identificar anticuerpos específicos han sido hemaglutinación e inmunocitoquímica indirecta. En el suero de pacientes con desórdenes endócrinos, son reconocidos anticuerpos hacia múltiples tejidos endócrinos. No es claro si estas múltiples reacciones indican la presencia de múltiples poblaciones de anticuerpos a múltiples antígenos o la presencia de un antígeno inmunogénico particular en múltiples tejidos endócrinos (11). En el suero de pacientes con FOP han sido detectados anticuerpos hacia corteza adrenal , tiroides, mucosa gástrica, testículo, células del islotes pancreático y piel (8).
· Medida de anticuerpos anti-ovario
Vallotton y Forbes(45), usando citoplasma de ovocito de conejo como antígeno, fueron los primeros en reportar anticuerpos antiovario en 4 de 45 pacientes con FOP y en 1 de 45 controles usando técnicas de inmunofluorescencia indirecta.
Inmunoglobulinas circulantes que se unen a proteínas ováricas han sido identificadas por inmunocitoquímica, inmunofluorescencia indirecta (IFI), tinción con inmunoperoxidasa (que permiten localizar la unión de un anticuerpo a un compartimiento en particular), Dot blot y recientemente Western blot. Las estructuras celulares antigénicas reportadas incluyen folículos desarrollados en mayor medida que folículos primordiales, oocitos, cuerpo lúteo, teca y granulosa. En la mayoría de los estudios de referencia algunos pacientes con FOP tienen anticuerpos antiovario mientras muy pocos sujetos controles presentan estos anticuerpos (8).
La inmunofluorescencia es una técnica que consume tiempo y es difícil de replicar. Es por ello que algunos investigadores diseñaron Elisas (enzimoinmunoensayos) usando variadas proteínas de ovario. Aunque es un ensayo más fácil de realizar ocasiona confusión(2). Pocos investigadores han intentado identificar anticuerpos antiovario usando técnicas bioquímicas como Radioinmunoensayos(Ria) y/o Elisas(5)(10)(17)(18). Coulam y Ryan (7) reportaron que pacientes con FOP tienen incrementado unión de inmunoglobulinas circulantes a proteínas iodadas, solubilizadas, extraídas de ovario de mujeres premenopaúsicas normales, así como fueron detectadas en pacientes con desórdenes endócrinos asociados. Sin embargo esta técnica se ha dejado de usar debido a sus resultados contradictorios.
· Autoanticuerpos en FOP con autoinmunidad adrenal
1-Ant célula esteoidea
2-Ant citoplasma adrenal
El descubrimiento en la década del 70 de los autoanticuerpos hacia la corteza adrenal (anticitoplasma adrenal) dio un importante ímpetu al estudio de la naturaleza autoinmune de la enfermedad de Addison idiopática. Dos variedades de anticuerpos adrenales fueron subsecuentemente reconocidos en el suero de pacientes con enfermedad de Addison usando IFI sobre secciones de glándulas adrenales humanas o provenientes de monos. Una variedad demostró solamente reactividad contra todas las capas de la corteza adrenal, mientras la otra variedad también reaccionó contra antígenos citoplasmáticos de otras células productoras de esteroides presentes en el ovario, testículo y placenta (teca interna del folículo ovárico, célula de Leydig de testículo, sinciotrofoblasto placentario). Irvine y col.. (46) fueron los primeros en demostrar anticuerpos dirigidos hacia la célula productora de esteroides. Realizando IFI con preparaciones de placenta, adrenal y ovario los citados autores encontraron que el suero de pacientes addisonianos reaccionaba con los tres tejidos. Esta última variedad de anticuerpos fue llamada anti-células esteroideas y su reactividad pudo ser absorbida por homogenatos adrenales, confirmando la reacción cruzada con el anticitoplasma adrenal(21). El título de anticuerpos de pacientes con sólo autoanticuerpos adrenales no es afectado por estudios de absorción con tejido gonadal. En contrapartida, el anticuerpo anticélula esteroidea está dirigido hacia determinantes antigénicos encontrados en todas las células productoras de hormonas esteroides(21). Hay una absoluta asociación entre la presencia de anticélula esteroidea y el anticitoplasma adrenal: el anterior sólo es detectable cuando este último está presente (16)(21). El anticuerpo anticélula esteroidea es del tipo IgG y se une dentro del ovario a células del hilio, células de folículos desarrollados así como células de la granulosa y de la teca y células del cuerpo lúteo. No es tejido ni sexo específico.
El anticuerpo anticélula esteroidea no fue encontrado en un estudio entre pacientes sin anticuerpos adrenocorticales o en pacientes con insuficiencia ovárica sin enfermedad de Addison (325 sujetos normales, 21 con síndrome de Turner, 505 con diabetes mellitus insulino dependiente, 15 falla ovárica idiopática, 35 con enfermedad de Addison) (21).
Casi todos los pacientes con amenorrea primaria y enfermedad de Addison tienen un título detectable de anticélulas esteroideas; el 60% de los pacientes con amenorrea secundaria y enfermedad de Addison muestran estos anticuerpos, pero los mismos no pueden ser detectados en pacientes con disgenesia gonadal, con anormalidades cromósomicas, falla ovárica no asociada a enfermedad Addison o controles normales (Tabla 8). En el seguimiento de pacientes addisonianos aproximadamente el 40% de las mujeres desarrollan falla ovárica en un período de 10 a 15 años, mientras en los hombres la presencia de anticuerpos anticélula esteroidea no anuncia falla gonadal.
La sensibilidad/ especificidad/ valor predictivo de los anticuerpos anti-células esteroideas en predecir la falla ovárica, en mujeres con SPA tipo I quienes inicialmente tenían función adrenal y ovárica normal fue de 100, 56, 50 % respectivamente (2)(4). La mayoría de los pacientes con SPA tipo I poseen anticuerpos anticélula esteroidea que correlaciona fuertemente con el hipogonadismo(21). Confirmando hallazgos anteriores de Irvine (1975) y Ruehsen (1972).
La mera presencia del anticuerpo en el suero de un paciente no es ciertamente evidencia para el significado patogénico del anticuerpo. El autoanticuerpo puede ser también evidencia de destrucción celular. Ha sido demostrado que el suero de pacientes con SPA tipo I y enfermedad de Addison (positivo para anticélulas esteroideas y anticitoplasma adrenal) conteniendo altos títulos de anticuerpos es citotóxico para células granulosas en cultivo en presencia de complemento. La citotoxicidad dependiente de complemento de los anticuerpos anti-células estroideas puede ser uno de los mecanismos que conducen a la destrucción de células productoras de esteroides in vivo y así a la falla ovárica.
Tabla 8 Prevalencia de anticuerpos anticélula esteroidea (4)
Prevalencia de anticuerpos anticélula esteroidea
Falla ovárica
Amenorrea/Infertilidad menos del 1 %
Con enfermedad tiroidea autoinmune o DM tipo I 5-10%
Con enfermedad de Addison
--amenorrea primaria 100%
--amenorrea secundaria 60%
Enfermedad de Addison sin falla ovárica
Casos aislados 10-20%
Con hipotiroidismo/ candidiasis (SPA tipo I) 60-80%
Con enfermedad tiroidea autoinmune 25-40%
SPA tipo I sin enfermedad de Addison 10%
Enfermedades tiroidea autoinmune o DM tipo I menos del 1%
Antígenos específicos de los anticuerpos anticélula esteroidea
· citocromo p450-c21 (21 hidroxilasa)
· citocromo p450-c17 (17 hidroxilasa)
· citocromo p450-scc (col.esterol desmolasa)
En los años recientes, se ha progresado en la identificación
del antígeno blanco de los anticuerpos anti-citoplasma adrenal
y anti-células esteroideas. Ha sido demostrado que el citocromo
p450-c21 (21 hidroxilasa: enzima que cataliza el paso de 17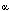 OH
progesterona y progesterona a 11-deoxicortisol y deoxicorticosterona)
es el mayor autoantígeno reconocido por autoanticuerpos presentes
en pacientes con enfermedad de Addison, ya sea en la enfermedad adrenal
aislada(29) o en la asociada a hipotiroidismo, SPA tipo II, también
OH
progesterona y progesterona a 11-deoxicortisol y deoxicorticosterona)
es el mayor autoantígeno reconocido por autoanticuerpos presentes
en pacientes con enfermedad de Addison, ya sea en la enfermedad adrenal
aislada(29) o en la asociada a hipotiroidismo, SPA tipo II, también
(dependiendo del autor) en SPA tipo I. Esto ha sido confirmado en estudios realizados en pacientes addisonianos, con la enzima nativa, producida en Escherichia col.i, levaduras, líneas celulares y traducción/transcripción in vitro(27)(37) .
En el SPA tipo I se ha postulado que el anticuerpo esté dirigido
hacia otro miembro de la familia de los citocromos p450 ( p450-scc y p450-c17)
y hacia una proteína de 51 kDalton (29). La citocromo p450-scc
es la enzima mitocondrial que cliva la cadena lateral del col.esterol,
en el pasaje de col.esterol a pregnenolona y la p450-c17 (17 hidroxilasa)
enzima microsomal que convierte pregnenolona y progesterona en 17OH pregnenolona
y 17OH progesterona respectivamente. Se ha sugerido que ambos citocromos
son el mayor componente del anticuerpo anticélula esteroidea. La
aparición de los autoanticuerpos usualmente precede a la falla
ovárica. Sin embargo no todos los investigadores han podido confirmar
la presencia de estos autoanticuerpos. De las enzimas esteroidogénicas
la p450- c21 es específica de adrenal, la p450-c17 está
expresada en ambos tejidos adrenal y gonadal mientras la p450- scc está
presente en adrenal, gónada y placenta (16). La proteína
de 51 kDa está presente en islotes pancreáticos, células
granulosas y placenta. Es posible que para algunos autoanticuerpos anti-célula
esteroidea falten identificar los antígenos productores.
En el caso de los autoanticuerpos a enzimas esteroideogénicas muchos estudios recientes han sido conducidos sobre la distribución de estos autoanticuerpos en diferentes grupos de pacientes.
En el SPA tipo I hay algunas discrepancias.
En 1995 Winqvist y col.(29). seleccionaron pacientes con insuficiencia adrenal y signos asociados de autoinmunidad gonadal para identificar los autoantígenos responsables de la reactividad de autoanticuerpos comunes hacia células esteroideas. El suero de la mayoría de los pacientes con enfermedad de Addison y de todos los pacientes con SPA tipo I reconocieron la citocromo p450-scc humana expresada en bacterias por Western blot. Cuando se empleó como sustrato para Western Blot el tejido de órganos productores de esteroides (placenta, células granulosas y células de Leydig) la enzima fue reconocida en todos los tejidos por los pacientes con SPA tipo I y por cinco de trece pacientes con enfermedad de Addison. Los restantes mostraron reactividad en uno o dos tejidos. Sólo dos pacientes fallaron en identificar la cit p450-scc extraída de tejido aunque ambas identificaron a la enzima expresada en bacterias.
Una razón para la variabilidad en la identificación de la citocromo p450-scc en diferentes preparaciones antigénicas puede ser que la enzima expresada en bacterias y la derivada de células y tejidos provean diferentes epitopes conformacionales. La mayoría de los autoanticuerpos reconocen epitopes formados por el plegado tridimensional del autoantígeno (conformacionales). Es importante reconocer que los autoantígenos y fragmentos de autoantígenos producidos en bacterias frecuentemente son incapaces de plegarse de la misma forma, por ende los resultados obtenidos con los mismos deben ser interpretados con precaución. De este modo, las preparaciones de dos especies diferentes podrían explicar diferente reactividad.
La inhibición de la actividad enzimática implica que algunos epitopes pueden estar cerca del sitio catalítico y/o sitios de unión del sustrato a la enzima.
La discrepancia en los distintos reportes sobre la distribución de los distintos autoanticuerpos hacia estos autoantígenos (p450-scc, p450-c17 y p450-c21) entre pacientes con enfermedad de Addison y SPA tipo I estarían relacionadas, al menos en parte, al origen del autoantígeno usado para detectar estos autoanticuerpos.
Un nuevo autoantígeno gonadal con un aparente peso molecular de 51 kDalton fue identificado por la mayoría de los sueros con enfermedad de Addison en una fracción placentaria humana y en células de la granulosa humana. Este presenta una movilidad separada de las proteínas identificadas en Western blot, por antisueros específicos dirigidos contra la p450-scc, p450-c17 y aromatasa. Con el uso de microscopio confocal fue encontrada una diferente localización subcelular, dentro del retículo endoplásmico, similar a la 21 hidroxilasa. El peso molecular puede indicar que esta enzima es otro autoantígeno de la familia del citocromo p450 (29) . El primer reporte que indicaba que el suero de pacientes con SPA tipo I inmunoprecipitaba una proteína de 51 kDa en lisados de células de islotes pancreáticos de ratas marcados con 35S, sin reacción en el suero de pacientes con diabetes mellitus insulino dependiente, enfermedad de Addison u otro desorden autoinmune fue hecho por Velloso y col. (1994).
En otro trabajo, Winqvist y col.. (25) encontraron que todos los pacientes con SPA tipo I identificaban la p450-scc en Western blot de fracciones mitocondriales adrenales bovinas, en Western blot de la enzima expresada en bacterias y en inmunoprecipitación de lineas celulares adrenocorticales humanas radiomarcadas. Esta enzima ha sido clonada y su gen está localizado en el brazo corto del cromosoma 15. Su peso molecular calculado es de 54 kDa. La presencia de autoanticueros contra esta enzima en el SPA tipo I ha sido confirmada por otros autores (Uibo y col.) (37). Song y col. (32) no han encontrado autoanticuerpos hacia p450-scc cuando esta fue producida en bacterias.
Es interesante el hecho que el suero de pacientes con SPA tipo I parecería no inhibir la actividad enzimática de la p450-scc.
En este estudio no parecen ser detectados estos autoanticuerpos en el suero de pacientes con enfermedad de Addison autoinmune. Sin embargo en estudios posteriores sí han sido detectados por los mismos autores (29).
En este trabajo no encontraron autoanticuerpos dirigidos hacia la 21 hidroxilasa en el suero de pacientes con SPA tipo I, sin embargo otros laboratorios han mostrado claramente que el 50% de los pacientes con SPA tipo I posee estos autoanticuerpos en el suero.
Algunos autores no han detectado reactividad hacia la 17 hidroxilasa
en el suero de pacientes con SPA tipo I (Winqvist y col...25) Song y col..(32))
por Western blot usando fracciones mitocondriales y microsomales adrenales
(nativas), mientras que mostraron suave reactividad cuando las mismas
fueron expresadas en bacterias. Mientras otros autores (Krohn y col..
(37)) han reportado en la mitad de los pacientes con SPA tipo I la presencia
de autoanticuerpos hacia la 17hidroxilasa usando
el antígeno producido en bacterias y nativo. Otros autores han
reportado que los mismos pueden ser inmunoprecipitados en el 30% de los
sueros de pacientes con el mencionado síndrome(37).
En 1992 Winqvist y col.. reportaron que el mayor autoantígeno en la enfermedad de Addison idiopática de comienzo en la adultez es la 21 hidroxilasa.
Algunas de las discrepancias sobre la ocurrencia de los autoanticuerpos entre las distintas publicaciones puede deberse a las distintas preparaciones de autoantígenos y a los métodos de detección empleados. En el caso de la 21 hidoxilasa estudios en los cuales la longitud total de la molécula de 21 hidroxilasa y una molécula modificada fueron expresados en sistemas de traducción/transcripción in vitro han indicado que el sitio de unión del anticuerpo parece ser dependiente de la cooperación entre la parte central y carboxilo terminal de la molécula. Consecuentemente, los fragmentos de 21 hidroxilasa expresados en bacterias y usados en Western blot podrían no reaccionar bien con los autoanticuerpos presentes en algunos sueros. Song y col..han confirmado la importancia de la porción carboxilo terminal de la molécula para el sitio de unión del anticuerpo, en un estudio usando fragmentos de 21 hidroxilasa expresados en bacterias (32). La importancia de esta región ha sido enfatizada en estudios en los cuales la enzima contenía una única mutación en un aminoácido. En particular, la mutación que ocurre naturalmente de prolina 453 (Pro453) a serina resulta en una marcada reducción en la capacidad del autoanticuerpo en reconocer la enzima, pero en escaso efecto sobre la unión del anticuerpo producido en conejos. La sustitución de este aminoácido está relacionada a una dismuída actividad enzimática en la hiperplasia adrenal no clásica.
Una situación similar puede ocurrir en el caso de los sitios de
unión para los autoanticuerpos hacia p450-c17 y p450-scc, donde
los epitopes reactivos con autoanticuerpos parecerían ser conformacionales(16).
En vista de estos problemas Chen y col.. (16)(1996) determinaron la presencia
de autoanticuerpos a enzimas esteroideogénicas: 17
hidroxilasa cit p450-scc y 21 hidoxilasa usando un ensayo de inmunoprecipitación
basado en autoantígenos producidos por sistemas in vitro de traducción/transcripción
marcados con 35S que parecería tienen mayor ventaja sobre los fragmentos
producidos en bacterias. Similares ensayos han sido usados para GAD (glutámico-aspártico
decarboxilasa) mostrando ser más sensibles y específicos.
De su estudio se desprende que:
1-el anticuerpo anti- 17ahidroxilasa ocurre frecuentemente en pacientes
con SPA tipo I (55%) SPA tipo II (33%), y pacientes con anticitoplasma
adrenal (ACA) positivo sin enfermedad de Addison, mientras se encuentra
con menor frecuencia en pacientes con enfermedad de Addison aislada (5%)(tabla
9). Sólo un paciente con FOP aislada tuvo anticuerpos detectables
y fue en muy baja concentración. Estos datos están de acuerdo
con algunos autores mientras otros sugieren una presencia o ausencia directamente
en pacientes con SPA tipo I(29).
Tabla 9 Porcentaje de autoanticuerpos presentes (16)
| Anticuerpos |
SPA tipo I | SPA tipo II | ACA (+) sin Addison | Addison | FOP aislada |
| p450-c21 |
64% |
96% |
86% |
64% |
0/17 |
| p450-c17 | 55% | 33% | 20% | 5% | 0/17 |
| p450-scc | 45% | 42% | 20% | 9% | 0/17 |
| ACA | 73% | 87% | 100% | 82% | 0/17 |
| ACE | 45% | 36% | 21% | 6% | 0/17 |
ACA: anticitoplasma adrenal
ACE: anticélula esteroidea
2-La p450-scc fue encontrada predominantemente en pacientes con SPA tipo I y SPA tipo II( 45 y 42% respectivamente) Además sólo el 20 % con ACA (+) sin enfermedad de Addison fueron positivos. La ocurrencia de este anticuerpo en enfermedad de Addison aislada fue del 9% y no se encontró en ningún paciente con FOP aislada. Winquist y col.. (29) detectaron p450-scc en todos los pacientes con SPA tipo I y en el 70 % de los pacientes con enfermedad de Addison. Otros autores reportaron 50% de positividad en pacientes con SPA tipo I, pero no encontraron reactividad en el SPA tipo II y en ninguno de los pacientes con enfermedad de Addison idiopática(16).
3-Los anticuerpos dirigidos hacia la p450-c21 fueron detectados en el 64% de pacientes con SPA tipo I, en el 96% de los pacientes con SPA tipo II, en el 64% de los pacientes con enfermedad de Addison aislada y en el 86% de los pacientes con ACA positivo sin enfermedad de Addison mientras no fueron detectados en pacientes con FOP.
Todos los sueros positivos para 17ahidroxilasa fueron también positivos para p450-c21 excepto un caso. Hay concordancia entre la presencia de ACA medido por IFI y 21 hidroxilasa medido por inmunoprecitación en todos los grupos de pacientes estudiados (tabla 10). Esto indica que la 21 hidroxilasa es el mayor autoantígeno en la enfermedad adrenal autoinmune sin reparar si se presenta aislada o formando parte de un síndrome.
Tabla 10 Resultados comparativos entre anticitoplasma adrenal (ACA) determinado por inmunofluorescencia (IF) y 21 hidroxilasa (21OH) determinado por inmunoprecipitación (IP) en el suero de 108 pacientes.
| ACA(+) IF(+) | ACA(-) IF(-) | |
| 21 OH IP(+) | 88 | 3 |
| 21 OH IP(-) | 10 | 7 |
Los autoanticuerpos a 17 hidroxilasa y p450-scc parecerían ser los mayores componentes de los anticuerpos anticélula esteroidea por IFI.
No se demostró la presencia de ninguno de los tres anticuerpos (p450-c17, p450-c21, p450-scc) en pacientes con FOP aislada. Excepto un solo caso bajos niveles de p450-c17. Esto concuerda con los trabajos reportados por Wheatcroft y col..(10) y con estudios realizados por IF que muestran que los anticuerpos anticélula esteroidea no son usualmente encontrados en pacientes con FOP aislada. (16)
Para destacar es el hecho que estos autoantígenos arriba mencionados son enzimas intracelulares como en muchos otros casos de enfermedades autoinmunes así como la peroxidasa en la tiroiditis autoinmune, la Na+K+ ATPasa en la gastritis autoinmune , la glutamato decarboxilasa en la diabetes mellitus. La predilección de enzimas como autoantígenos es una intriga.
Aparte de los autoanticuerpos hay otros argumentos que consideran la falla ovárica con anticélulas esteroideas positivo como una enfermedad autoinmune: la histología de la lesión ovárica.
· Autoanticuerpos en pacientes con FOP sin autoinmunidad adrenal
1-Autoanticuerpos a órganos endócrinos
La más prevalente autoinmunidad endócrina reportada en pacientes con FOP sin autoinmunidad adrenal es la autoinmunidad tiroidea (14%), seguida por la presencia de anticuerpos anticélula parietal (4%), DMID (2%) y miastenia gravis o positividad para anticuerpos hacia el receptor de acetilcol.ina (2%).
Tabla 11 Perfil de anticuerpos en pacientes con FOP idiopática sin autoinmunidad adrenal (8)
| FOP | Enf.tiroidea | Anticél.parietal | ICA/DMID | Antirecep Ach | Antic No órgano específ.: |
| 629 | 86 (13%) | 23 (3.7%) | 11 (1,7%) | 14 (2,2%) | varios |
Sin embargo la prevalencia general de positividad para anticuerpos tiroideos y anticélula parietal gástrica es sólo ligeramente superior a la prevalencia encontrada en la población normal. La miastenia gravis o DMID, ambas enfermedades autoinmunes relativamente poco comunes, son encontradoas con mayor frecuencia en pacientes con FOP ( 2-4%). En LES, anticuerpos antinúcleo factor reumatoideo también han sido reportados con mayor frecuencia de lo normal en pacientes con POF. La relación del LES y FOP está fortalecida por el hallazgo de la presencia de anticuerpos antiovario en el 84% de los casos de pacientes femeninas con LES.
2-Anticuerpos antiovario
Una fuerte evidencia a favor de la naturaleza autoinmune de la FOP aislada es la presencia de anticuerpos dirigidos hacia estructuras ováricas en el suero de estos pacientes (5)(7)(8)(10)(17)(18)(48)(52).
Los anticuerpos anticitoplasma ováricos han sido descriptos desde 1968 por Irvine y col. seguidos posteriormente por Moraes-Rueshen y col.. Ambos autores usaban técnicas de IFI para su detección. Los anticuerpos fueron encontrados en sólo la mitad de los pacientes con FOP con enfermedad autoinmune asociada, los pacientes sin síntomas autoinmunes fueron negativos. Sotsiou y col. extendieron estos estudios y encontraron que los anticuerpos anticitoplasma ovárico eran detectados en una minoría de los pacientes FOP y sólo en aquellos con anticitoplasma adrenal positivo. Anticuerpos ováricos distintos de los anticélula esteroide han sido detectados en pacientes con FOP sin otra evidencia de enfermedad autoinmune, (17) pero su prevalencia y papel en la patogénesis permanece incierta debido a las diferencias en las poblaciones estudiadas y a las técnicas usadas. Wheatcroft y col.(10) examinaron el suero de 45 pacientes con FOP (5 iatrogénicas, 9 asociadas a otra enfermedad autoinmune, 27 con FO idiopática) así como 4 pacientes con infertilidad por síndrome de Turner y 41 mujeres pre y posmenopaúsicas como controles. Ellos encontraron autoanticuerpos en pacientes con FOP por Elisa, usando preparaciones de antígenos ováricos humanos . Las preparaciones de ovario variaban ampliamente en su capacidad de unión. Al menos una proporción de estos anticuerpos no eran específicos pues reaccionaban con otros antígenos como los obtenidos de trompas de Fallopio. Además los anticuerpos también se encontraban en pacientes con anormalidades cromosómicas y pacientes con falla ovárica iatrogénica por radiación o quimioterapia. Esto último puede ser debido al trauma y a la liberación de antígenos asociados con este proceso. Ha sido reportado previamente la presencia de anticuerpos anti ovario en pacientes con disgenesia gonadal (10).
Recientemente Luborsky y col.(17) empleando una técnica de Elisa encontraron que aproximadamente el 70% de los sueros de pacientes con FOP contenían anticuerpos a ovario u oocitos. Datos más tempranos usando inmunohistoquímica sugerían que estos anticuerpos ocurrían en el 18 a 60% de los pacientes con FOP. La variación en la reactividad entre preparaciones ováricas puede ser debido a variaciones cíclicas en las proteínas antigénicas.
Estos anticuerpos se piensa que interfieren con el desarrollo folicular por su interacción con la célula productora de esteroides. En esta interacción, el complemento puede ser activado seguido por la atracción de leucocitos y la destrucción de la célula ovárica (33) .
La IFI ha sido la técnica comúnmente usada para identificar anticuerpos ováricos, pero variaciones en el origen del tejido ovárico empleado (humano, primate,etc) y en el estadío de maduración del mismo para la detección de anticuerpos humanos ha sido cuestionable. Este es particularmente importante debido a que el desarrollo de estructuras ováricas está influenciado por el ciclo menstrual. Aún usando tejido ovárico humano la incidencia determinada por IFI fue de 2 a 50%.
Wheatcroft y col.. (17) realizaron un estudio comparativo entre IFI/Elisa para comparar los resultados obtenidos con ambas metodologías.(18) Se examinaron la detección de anticuerpos antiovario en tres grupos de pacientes: un grupo con FOP idiopática, un grupo con enfermedad autoinmune asociada y otro grupo de pacientes con IFI positiva, de las cuales tres desarrollaron luego falla ovárica. Sólo tres de 42 pacientes del primer grupo (idiopática) tenían IFI positiva sobre secciones de ovario de mono, aun cuando el anticuerpo detectado fue en parte nuclear. Ninguno de ellos fue positivo por Elisa. En contraste, la frecuencia de pacientes positivo por Elisa para el tercer grupo (IFI positivo) fue del 53%. En reportes previos se ha descripto un mayor acuerdo entre ambas metodologías usando ovario humano como fuente de tejido (18).
La presencia de anticuerpos antiovario es considerada como un marcador para identificar la participación de un rol inmunológico en el mecanismo de la FOP. Sin embargo problemas con las técnicas y la interpretación de los resultados ( inmunohistoquímica, IFI) , anticuerpos positivos encontrados post cirugía (Elisa) (17), seguido a la recuperación de oocitos para fertilización in vitro y aún en síndrome de Turner (10) hacen que el real significado de los mismos sea cuestionable.
Fénichel no encontró en su estudio sobre anticuerpos antiovario relación entre la duración de la amenorrea y la presencia de los mismos, pudiendo ser detectados luego del comienzo de la menopausia. Se ha reportado disminución de su título luego de la terapia con estrógenos (5). La ausencia de anticuerpos antiovario en una amplia proporción de pacientes con FOP usando Elisa o IFI sugiere que el anticuerpo no es un mediador inicial de la enfermedad, siendo los linfocitos T probablemente cruciales en la patogénesis.
Empleando inmunoblot hacia dos antígenos ováricos microsomales Wheatcroft y col.. (1997) no fueron capaces de demostrar un patrón consistente de unión ya sea para el grupo de pacientes con FOP idiopática o autoinmune (18) .
Charreau y col. (52) en estudios previos mediante Dot blot encontraron 79/244 (33%) pacientes con FOP reacción positiva contra proteínas de ovarios humanos (Tabla 12). En un trabajo posterior (1998) con 54 sueros de pacientes con FOP, en los cuales había sido encontrado por la metodología anteriormente citada anticuerpos antiovario, y 36 controles analizados por Western blot utilizando como antígenos proteínas citosólicas depletadas de IgG y proteínas de placenta y de músculo como controles, encontraron sólo en el 11% de los sueros evidencia de un antígeno específico de peso molecular 55kDa(51)(52).
Tabla 12 Distribución de anticuerpos anti-receptor de FSH y antiovario en pacienes con falla ovárica prematura, incluida SOR(48)
| Nro de Pacientes | Gonadas | Edad última menstruación | Embarazos previos | Otras enfermedades | Anti-receptor FSH | Anti-ovario |
| 10 | Ovarios con folículos primordiales | Amenorrea primaria a 31 años | 2 G1 P1 8 G0 P0 |
8 Miastenia 1 Artritis |
10(100%) | 9(90%) |
| 10 | -- | 16 - 36 años | 10 G0 P0 | Miastenia - Artritis - Tiroiditis | 10(100%) | -- |
| 48 | Ovarios hipoplasicos sin folículos | 17 - 30 años | 48 G0 P0 | 12 enf. Tiroideas 2 Vitiligio 6 Poliquistosis 2 LSE |
0 | 14(29.2%) |
| 176 | -- | Amenorrea Primaria a 36 años | 176 G0 P0 | 40 enf. Tiroideas 2 Vitiligio 12 Poliquistosis de ovario 5 Artritis 1 Addison 1 Diabetes mellilus |
0 | 56(31.8%) |
| Total=244 | -- | -- | -- | -- | 20(8.2%) | 79(32.4%) |
Para poder determinar la relevancia diagnóstica de los anticuerpos hacia el ovario en pacientes con FOP autoinmune e idiopática son requeridas futuras caracterizaciones.
3-Autoanticuerpos hacia receptores (síndrome de ovario resistente)
Anticuerpos que bloquean el receptor han sido descriptos como causales de miastenia gravis (anticuerpos bloqueantes del receptor de acetilcol.ina), algunas formas de insulino resistencia (anticuerpos bloqueantes del receptor de insulina), y enfermedad de Graves (anticuerpos estimulantes del receptor de TSH). Es fácilmente entendible que el receptor de FSH o LH pueda ser el blanco para anticuerpos bloqueantes. Experimentos mostrando una interacción de anticuerpos con estos receptores en pacientes con FOP han sido descriptos por varios autores(15)(33)(51) (tabla 13). Datos inconsistentes fueron generados con respecto a la prevalencia y al blanco exacto de estos anticuerpos.
El receptor de LH no parece ser el antígeno en la FOP desde que Austin y col.aboradores fueron incapaces de detectar anticuerpos que inhibieran la unión de HCG marcada con yodo 125 al cuerpo lúteo de rata o humano. Otros enfocan la atención en intentar detectar anticuerpos al receptor de FSH ya que la presentación clínica e histopatológica en la FOP es consistente con la falla del folículo de responder a la FSH. Hay evidencia de un inhibidor de la unión de FSH al receptor testicular de rata en el suero de pacientes con miastenia gravis y amenorrea hipergonadotrófica. Charreau, Chiauzzi y col.(30) han demostrado que este pacientes con el síndrome de resistencia del ovario tenían inmunoglobulinas IgG circulante que inhibían la unión de FSH a su receptor.
En una serie de 244 mujeres con FOP con cariotipo normal, sólo encontraron 20 casos (8,1%) con anticuerpos circulantes contra los receptores para FSH. Todas estas pacientes, con folículos ováricos arrestados en el estadío de folículos primordiales y con una falta de respuesta al tratamiento con gonadotrofinas, cumplían con el criterio de síndrome de ovario resistente y nueve de ellas tenían miastenia gravis asociada (Tabla 12 ). Los autores sugieren que el desarrollo de anticuerpos dirigidos contra el receptor de gonadotrofinas o hacia un dominio de la membrana relacionado al receptor explicaría la incapacidad del ovario de responder a la acción trófica de la FSH endógena o al estímulo exógeno con dosis masivas de gonadotrofinas para tratar de inducir la ovulación, (15)(27)(30) contribuyendo al cuadro clínico de la hipofunción. Si estos hallazgos y los de otros grupos pueden confirmarse, podría evitarse la biopsia en el diagnóstico del SOR.
En 1991 van Weissenbruch, Hoeck y col.. (33) demostraron la presencia de una inmunoglobulina de tipo IgG en el suero de 21 de 26 pacientes con FOP estudiadas, capaz de bloquear, in vitro, la síntesis de DNA inducida por FSH de la célula de granulosa. Estos pacientes fueron caracterizados como Síndrome de ovario resistente. Estos anticuerpos no tienen efecto sobre la esteroideogénesis inducida por LH. Confirmando los hallazgos de Chiauzzi y col..
Algunos autores (Anasti y col..) critican que todos los trabajos empleaban receptores de gonadotrofinas no humanos (tabla13), sufriendo de un potencial defecto en el diseño experimental. Datos recientes usando receptores de LH y FSH clonados indican que el receptor de gonadotrofinas es altamente selectivo para su ligando humano. Anasti y col.. usando estos receptores no pudieron demostrar la presencia de inmunoglobulinas dirigidas contra las gonadotrofinas o sus receptores en pacientes con FOP. Estos datos no concuerdan con los reportados por Charreau y col.., quienes demostraron la presencia de inmunoglobulinas bloqueantes del receptor de FSH en veinte pacientes con Síndrome de Savage. Estos anticuerpos no fueron encontrados en controles ni en mujeres con amenorrea hipergonadotrófica de otro origen o enfermedades autoinmunes. Una característica derivada del análisis cinético de los datos de unión para la hormona y su receptor se puede observar en la figura 2. Los resultados de dicho análisis, obtenido con cantidades variables de inmunoglobulina y estudiando la unión de 125I-FSH a receptores de membrana, representados según el gráfico de Scatchard, demuestran que los pacientes con SOR pueden subdividirse en dos grupos. Uno de los grupos poseería inmunoglobulinas que se unirían al receptor o a dominios de membrana relacionados con el receptor, con una afinidad aparente de 10-12M-1, mostrando una inhibición del tipo “irreversible”. Esto podría explicar por que la resistencia ovárica no puede sobrepasarse con niveles elevados de FSH circulante in vivo. Un segundo grupo de pacientes, mostraría un tipo de inhibición “reversible” con valores de afinidad para el anticuerpo similares a la constante de afinidad aparente para la FSH y sus sitios receptores. Es posible que estas pacientes, expuestas a terapias con dosis altas de gonadotrofinas exógenas, puedan ocasionalmente responder e incluso ovular (48).
Figura 2 Inhibición de la unión de 125I-FSH a sus receptores específicos por inmunoglobulinas de pacientes con síndrome de ovario resistente.(48)
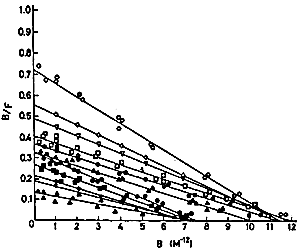
0-0 unión de FSH; otros signos : competición con IgG de pacientes con SOR.
Algunos investigadores han reportado un inhibidor de la unión de FSH (no inmunoglobulina) aislado del suero de pacientes con FOP. Esta molécula de bajo peso molecular efectivamente bloquea la acción de FSH a su receptor (14). La importancia de estos hallazgos necesita de futura elucidación.
Debe tenerse en cuenta que aunque este tipo folicular de disgénesis ovárica no se encuentra frecuentemente en pacientes con FOP, en teoría es el único caso (al menos hipotéticamente) en el que al revertirse las condiciones inhibitorias de la función ovárica podrían recomenzar los ciclos. Se ha reportado estimulación ocasional del desarrollo folicular, con ovulación y embarazo con tratamiento combinado con estrógenos y progesterona (48).
Tabla 13 Anticuerpos bloqueantes del receptor de LH o FSH en pacientes con FOP idiopática.
| Referencia |
Año | Sistema de ensayo | Sustrato | FOP con AA | FOP sin AA | Controles / otra enf |
| Austin y col. | 1979 | ensayo de unión recept.LH/HCG | cuerpo lúteo humano/rata | N.D. | 0/14 | 0/14 |
| Chiauzzi y col. | 1982 | ensayo de unión receptor de FSH | membrana testicular rata | N.D. | 2/11 | 0/5 |
| Tang y Faiman | 1983 | ensayo de unión rec FSH/LH | membrana test.bovino | N.D. | 1/9 | 0/18 |
| Moncayo y col. | 1990 | ensayo de unión recept.LH/HCG | cuerpo luteo bovino | N.D. | N.D: |
11/35 esterilidad FIV,endomet. |
| van Weissenbruch y co | 1991 | síntesis de DNA por cél.granulosa inducida por FSH | folículo ovárico de rata | 4 de 4 | 17/22 | 0/13 |
| Wheatcroft | 1994 | ensayo de unión recept.LH/HCG | bovino | 2 de 2 | 7/33 | 4 de 8 |
| Anasti y col | 1995 | ensayo de unión rec FSH/LH |
recombinante humana | 0/1 | 0/37 | 0/14 |
| Charreau y col.4 | 1999 | ensayo de unión de rec de FSH | membrana testicular rata | 20 de 24 | N.D. | dato no disponible |
4-Anticuerpos antizona pelúcida
La zona pelúcida es la matriz acelular que rodea a los oocitos desarrollados y ovulados y es también detectable en los folículos atrésicos. Los autoanticuerpos hacia la zona pelúcida (ZP) han sido descriptos como causa de infertilidad en la mujer (2). En la mujer con infertilidad inexplicable estos anticuerpos han sido detectados en el 5,6% de los casos, mientras en los controles normales la positividad parece ser de sólo 1,7%(4). En modelos animales se ha observado que estos anticuerpos interfieren con el desarrollo folicular y su presencia conduce a depleción folicular y amenorrea. Grootenhuis y col. recientemente encontraron por Elisa 3 de 34 pacientes positivos para IgG dirigidos hacia la proteína ZP humana recombinante Sin embargo, tres de seis mujeres postmenopaúsicas fueron también positivas. Por lo tanto es probable que estos anticuerpos sean el resultado del daño folicular más que su causa.
· Estimulación hormonal y antigenicidad del ovario
A fines de la década del 80, se identificó un nuevo grupo de pacientes que presentaba anticuerpos antiovario. Este grupo correspondía a aquellos pacientes que habían recibido tratamientos con gonadotrofinas en programas de superovulación para fertlizaciones in vitro. Análisis secuenciales antes y durante el tratamiento mostraron una incidencia significativa de anticuerpos circulantes durante la estimulación hormonal. Esta asociación ha sido observada también en pacientes con falla ovárica oculta que fracasan a tratamientos con FIV (48).
Es reconocido que no todos los componentes del ovario son igualmente antigénicos, siendo los folículos maduros y el cuerpo lúteo los de mayor antigenicidad. Es posible entonces que el uso de gonadotrofinas para la estimulación pueda llevara autoinmunidad como resultado de la estimulación suprafisiológica del ovario, sugiriendo fuertemente una interrrelación dinámica entre el hipotálamo, la hipófisis y el sistema inmune (48).
· Evidencia histológica de ooforitis
Histología del ovario
· FOP combinada con autoinmunidad adrenal y/o enfermedad de Addison
En estudio sobre un total de 215 casos, todos los pacientes con FOP y anti-célula esteroideas positivo tenían ooforitis linfocítica y de la totalidad de casos reportados, el 78% presentó anticuerpos anticélula esteroidea positivo (4). El 11% de los pacientes con FOP tenían evidencia de ooforitis.
La apariencia microscópica del ovario con ooforitis linfocítica fue quística en el 50% de los casos (con quistes pequeños o grandes) o normal. Se ha postulado que la formación de quistes se debe a los altos niveles de gonadotrofinas de estos pacientes.
Con respecto al patrón de infiltración microscópica hay marcada similitud en los casos reportados de ooforitis linfocítica en los diferentes compartimentos del ovario. En la mayoría de los casos, los folículos primordiales no están afectados, tampoco la corteza del ovario. Son los folículos desarrollados los que están predominantemente infiltrados por células inflamatorias mononucleares. No hay un patrón claro de incrementada densidad de infiltración con más folículos maduros. Los folículos preantrales están rodeados por un pequeño borde de linfocitos y células plasmáticas, mientras los folículos mayores tienen progresivamente más densidad de infiltrado usualmente en la teca interna y externa. La capa granulosa está usualmente al margen en este proceso hasta que ocurre la luteinización del folículo.(22)
Cuando los quistes están presentes ellos están luteinizados con una infiltración leucocítica marcada en la pared del quiste y destrucción de la línea celular. Los folículos atrésicos y cuando están presentes, el cuerpo lúteo y el cuerpo albicans están infiltrados. El patrón de infiltración confirma que el principal blanco para el ataque autoinmune es la célula productora de esteroide. Un infiltrado moderado puede estar presente en la médula o en la región hiliar del ovario. Y sorprendentemente hay infiltrados perivascular y perineural en el hilio del ovario.
El análisis inmunohistoquímico de la ooforirtis limfocítica revela que las células inflamatorias están pricipalmente formadas por linfocitos T CD4 y CD 8 positivos con escasas células B junto a un amplio número de células plasmáticas. También se puede encontrar macrófagos y células NK. La célula plasmática principalmente secreta IgG, pero también IgA e IgM, lo cual implica producción local de autoanticuerpos ováricos.
La presencia de FOP y enfermedad de Addison y/o autoinmunidad adrenal (2-10% de los casos) es casi con certeza un desorden endócrino autoinmune. Esto está evidenciado por:
1-la presencia de autoanticuerpos a células productoras de esteroides
2- la caracterización de autoantígenos compartidos entre
las células productoras de esteroides adrenal y ováricas
3- el cuadro histológico del ovario (infiltrado linfoplasmocelular
particularmente alrededor de la célula productora de esteroides)
· FOP en ausencia de autoinmunidad adrenal y/o enfermedad de Addison
El cuadro histólogico del ovario en pacientes con FOP sin autoinmunidad adrenal es el siguiente: en un 60% de los casos de FOP se carece de folículos ováricos y en algunos casos se han encontrado ovarios fibróticos. En el otro 40% de los casos los folículos están presentes, el 10% de estos casos tienen numerosos folículos y probablemente representen síndrome de ovario resistente.
Los casos de ooforitis linfocítica difícilmente son encontrados en pacientes con POF en ausencia de autoinmunidad adrenal/ enfermedad de Addison, sin embargo se ha demostrado la presencia de inmunoglobulinas en ovarios afectados sin ooforitis usando IFI : en el 50% de los casos hay tinción de la pared vascular (IgA,IgM,o IgG) y en 30% las células del estroma y las células foliculares fueron positivas para inmunoglobulinas. Se ha postulado que los anticuerpos hacia el ovario pueden estar presentes sin alcanzar niveles séricos detectables o inducir inflamación local. De hecho la histología de la FOP en ausencia de autoinmunidad adrenal/ enfermedad de Addison no es de ayuda para soportar una patogénesis autoinmune. Esto también se aplica a la atrofia encontrada en la mayoría de los casos. Este fenómeno puede representar el estadío final de un proceso autoinmune dirigido contra estructuras del ovario.
A favor de esta hipótesis está la asociación a otras enfermedades autoinmunes reportadas con mayor frecuencia en pacientes con POF.
· Recuperación luego de terapia inmunosupresora
Evidencia adicional de la etiología autoinmune es provista por seis reportes que documentan el retorno de la función ovárica luego de la terapia inmunosupresiva o recuperación de una enfermedad autoinmune (31)(49). El tratamiento con corticosteroides mejoró tres casos de FOP: dos pacientes, uno con anticuerpos antiovario y otra con evidencia de un infiltrado linfocítico del ovario experimentaron retorno temporario de la menstruación seguido de cortos cursos de glucocorticoides. La tercer paciente prepúber con elevados niveles de gonadotrofinas comenzó su desarrollo prepuberal normal seguido a la terapia glucocorticoidea por enfermedad de Addison. Dos pacientes con miastenia gravis reanudaron su menstruación luego de terapia inmunosupresiva. La primera experimentó una mejora temporaria seguida a plasmaféresis, mientras la segunda luego de una timectomía terapeútica. Sin embargo la inmunosupresión no es exitosa en todos los casos(8). En la figura 3 se puede observar el incremento en la unión de FSH a su receptor luego de una timectomía en una paciente con SOR, la paciente normalizó su cuadro hormonal y reinició sus ciclos menstruales. En la figura 4 la disminución en la absorvancia (Elisa) del suero de un paciente con FOP luego de terapia inmunosupresora con coticosteroides, la paciente regularizó sus ciclos y quedó embarazada. (Gentileza Dr. Charreau)
Figura 3 Unión de FSH a su receptor antes y después de la timectomía en una paciente con SOR (48).
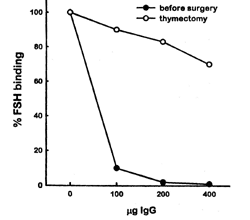
Figura 4 Cambio en concentración de anticuerpo antiovario en una paciente con FOP antes y después del tratamiento con corticosteroides (Elisa) (48)
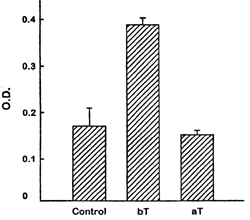
bT: antes del tratamiento
aT :después del tratamiento
OD:densidad óptica
Conclusiones:
· Aunque la presencia de anticuerpos específicos en el suero de pacientes, dirigidos hacia proteínas celulares de órganos afectados, sustenta el diagnóstico de enfermedad autoinmune, es bien conocido que el fenómeno de inmunidad celular o humoral necesita ser acompañado por enfermedad autoinmune demostrable fisiológica y clínicamente. De este modo, los anticuerpos circulantes son considerados marcadores inmunológicos de un epifenómeno más complejo.
· Si bien es cierto que no puede descartarse la posibilidad de una falla intrínseca en la maquinaria de las células blanco o un fenómeno de “down-regulation” del receptor en las células del ovario debido al elevado nivel gonadotrófico, la sóla presencia de anticuerpos contra el receptor de FSH permite explicar la resistencia hormonal de estos pacientes.
Bibliografía:
1-Kalantaridou S.N.; Davis S.R. and Nelson.L.M. Premature ovarian failure.
Endocrinol. Metabol. Clin. of North. Am. 1998; 27: 989-1006
2- Anasti N.A.,MD: Premature ovarian failure: an update. Fertil. and Steril.
1998; 70:1-15.
3-Rebar R.; Cedars I. M. Hypergonadotropic forms of amenorrhea in young
women. Endocrinol. Metabol. Clin. of North Am 1992:21:173-191.
4-Hoek A.,Schoemaker J and Drexhage A.: Premature ovarian failure and
ovarian autoimmunity. Endocrine Reviews 1997; 18: 107-134.
5-Fénichel P., Sosset C:, Barbarino-Monnier P., Gobert B., Hiéronimus
S., Béné M.C. and Harter M.: Prevalence, specificity and
significance of ovarian antibodies during spontaneous premature ovarian
failure. Hum. Reprod. 1997; 12: 2623-2628.
6-Wheatcroft N.J, Rogers C.A., Metcalfe R.A., Lenton E.A., Cooke I.D and
Weetman A.P: Is subclinical ovarian failure an autoimmune disease? Hum.
Reprod. 1997, 12: 244-249.
7-Coulam C and Ryan R.J.: Prevalence of circulating antibodies directed
toward ovaries among women with premature ovarian failure. Am. J. Reprod.
Immun.1985; 9: 23-24.
8-LaBarbera A., Miller M.M., Ober C, and Rebar R.W.: Autoimmune etiology
in premature ovarian failure. Am. J.Repod. Immun. 1988; 16:115-122.
9-Kim T.J.; Anasti J.N.; Flack M.R.; Kimzey L.M.; Defensor R.A. and Nelson
L.M. Routine endocrine screening for patients with karyotically normal
spontaneous premature ovarian failure. Obstetric and Gynecol.ogy 1997;
89: 777-779.
10-Wheatcroft N.J., Toogood A.A , Li T.C., Cooke I.D., Weetman A.P. Detection
of antibodies to ovarian antigens in women with premature ovarian failure.
Clin. Exp. Immunol. 1994;96:122-128.
11- Kutteh W.H. Immunology of multiple endocrinopathies associated with
premature ovarian failure. The Endocrinologist 1996;6:462-466.
12-Nelson L.M., Kimzey L.M., Merrian G.R. and Fleisher T.A. Increased
peripheral T lymphocyte activation in patients with karotypically normal
spontaneous premature ovarian failure. Fertil. Steril. 1991;55:1082-1087.
13-Rebar R.W., Erickson G.F., Yen SS: Idiopathic premature ovarian failure:
Clinical and endocrine characteristics. Fertil. Steril. 1982; 37:35-41.
14-Sluss P.M. and Schneyer A.L. Low molecular weight follicle-stimulating
hormone receptor binding inhibitor in sera from premature ovarian failure
patients. J. Clin. Endocrinol. Metab. 1992;74: 1242-1245.
15-Chiauzzi V., Cigorraga S., Escobar M., Rivarola M.A., and Charreau
E.H.: Inhibition of follicle-stimulating hormone receptor binding by circulating
immunoglobulins. J Clin Endocrinol and Metab1982;54:1221-1228.
16-Chen S., Sawicka J., Betterle C., Powell M., Prentice L., Volpato M.,
Rees Smith B., and Jadwiga Furmaniak. Autoantibodies to steroidogenic
enzymes in autoimmune polyglandular syndrome, Addison’s disease,
and premature ovarian failure. J.Clin. Endocrinol. Metab. 1996,81:1871-1876.
17- Luborsky J.L , Visintin I., Boyers S., Asari T, Dalswell B. and De
Cherney A. Ovarian antibodies detected by immobilized antigen immunoassay
in patients with premature ovarian failure. J. Clin. Endocrinol. Metab.
1990; 70:69-75.
18- Wheatcroft N.J., Salt C., Milford-Ward A., Cooke I.D., and Weetman
A.P. Identification of ovarian antibodies by immunofluorescence, enzyme-linked
immunoabsorbent assay or immunoblotting in premature ovarian failure.
Hum. Reprod. 1997; 12:2617-2622.
19- Aiman James, and Sementek Craig. Premature ovarian failure. Obstet.
Gynecol.. 1985; 66:9-14.
20- Coulam B. C., Adamsom S. C. and Annegers J.F. Incidence of premature
ovarian failure. Obstet. Gynecol.. 1986; 67:604-606.
21- Elder M., Maclaren N. and Riley W. Gonadal autoantibodies in patients
with hypogonadism and/or Addison´s disease. J. Clin. Endocrinol.
Metab. 1981; 52:1137-1142.
22-Coulam B.C. Premature gonadal failure. Fertil. Steril. 1982;38:645-655.
23-Conway G.B., Kaltsas G, Patel A.D. Characterization of idiopathic premature
ovarian failure. Fertil. Steril. 1996,65:337-341.
24-Olsen N.J., and Kovacs W.J. Gonadal steroids and immunity. Endocrine
Reviews 1996; 17:369-384.
25-Winqvist O., Gustafsson J., Rorsman F., Karisson A. and Kampe O. Two
different cytochrome p450 enzymes are the adrenal antigens in autoimmune
polyendocrine syndrome type I and Addison’s disease. J.Clin.Invest.
1993, 92:2377-2385.
26- Moncayo R. and Moncayo H. Autoimmunity and the ovary. Inmunol. Today
1992;13:255-258.
27-Lin Tan Seang, Hague W., Becker F., Jacobs H. Autoimmune premature
ovarian failure with polyendocrinopathy and spontaneous recovery of ovarian
follicular activity. Fertil Steril 1986; 45:421-425
28- Starup J and Sele V. Premature ovarian failure. Acta Obstet. Gynec.
Scand. 1973;52:259-268.
29-Winqvist O., Gebre-Medhin G., Gustafsson J.,Ritzen M.,Lundkvist Örjan
Karlsson A. and Kampe Olle. Identification of the main gonadal autoantigens
in patients with adrenal insufficiency and associated ovarian failure.
J. Clin. Endocrinol. Metab. 1995,80:1717-1723.
30-Escobar M.E., Cigorraga S., Chiauzzi V., Charreau E. and Rivarola M.
Development of the gonadotrophic resistant ovary syndrome in myasthenia
gravis: suggestion of similar autoimmune mechanisms. Acta Endocrinológica,
1982;99:431-436.
31-Blumenfeld Zeev,Sar’el Halachmi B.,Alik Peretz B.,Zehava Shmuel,Dov
Golan, Makler Amnom,Brandes J. Premature ovarian failure-the prognostic
application of autoimmunity on conception after ovulation induction. Fertil.
Steril. 1993;59:750-755.
32-Song Y-H, Connor E.L. Muir A. Autoantibody epitope mapping of the 21-hydroxilasa
antigen in autoimmune Addison’s disease. J. Clin. Endocrinol. Metab.
1994;78:1108-1112.
33-van Weissenbruch M., Hoek Annemieke, van VlietBleeker I., Schoemaker
J. and Drexhage H. Evidence for existence of immunoglobulins that block
ovarian granulosa cell growth in vitro. A putative role en Resistant Ovary
Syndrome. J. Clin. Endocrinol. Metab. 1991;73:360-367.
34-Nelson L., Anasti J. and Kimzey L., Defensor R.,Lipetz K., White B.,
Shawker T. and Merino M. Development of luteinized graafian follicles
in patients with kariotypically normal spontaneous premature ovarian failure.
J. Clin. Endocrinol. Metab. 1994;79:1470-1475.
35-Anasti J.N., Adams, Kimzey R, Defensor R, Zachary A and Nelson L.M.
Karyotycally normal spontaneous premature ovarian failure ; evaluation
of association with the class II mayor histocompatibility complex. J.
Clin. Endocrinol. Metab.1994;78:722-723.
36- Hill J, Welch W, Faris H and Anderson D. Induction of class II major
histocompatibility complex antigen expression in granulosa cells by interferon
gamma: A potential mechanism contributing to autoimmune ovarian failure.Am
J. Obstet Gynecol. 1990; 162: 534-540.
37-Uibo R., Aavik E., Peterson P. Autoantibodies to cytochrome P450 enzymes
P450scc, P450c17 and P450c21 in autoimmune polyglandular disease types
I and II and in isolated Addison’s disease. J.Clin.Endocrinol.Metab.
1994;78:323-328
38-Richardson J.S., Senukas V., Nelson J.F. Follicular depletion during
the menopausal transition: Evidence for accelerated loss and ultimate
exhaustion. J.Clin.Endocrinol.Metab 1987; 65:1231-1237.
39- de Moraes-Ruehsen M, Jones G.S. Premature ovarian failure. Fertil
Steril 1967;18:440-461.
40-Cameron IT., O’Shea F.C., Rolland J.M.,Hughes E.G., de Kretser
D.M. and Healy D.L. Occult ovarian failure: a syndrome of infertility,
regular menses, and elevated follicle-stimulating hormone concentrations.
J.Clin.Endocrinol.Metab. 1988;67:1190-1194.
41-Maxon W.S., and Col.ston Wentz A. The gonadotropin resistant ovary
syndrome. Seminars in Reproductive Endocrinology 1983;1:147-160.
42- van Campenhout J., Facog Vauclair R. and Maraghi K. Gonadotropin-Resistent
ovaries in primary amenorrhea. Obstet. Gynecol.. 1972; 40:6-11.
43-Jones G.S., de Moraes-Ruehsen M. A new syndrome of amenorrhea in association
with hypergonadotropism and apparently normal ovarian follicular apparatus.
Am. J. Obstet. Gynecol.. 1969; 104:597-600.
44-Immunology of multiple endocrinopathies associated with premature ovarian
failure. The Endocrinologist 1997, 34:156-161.
45-Vallotton M.B., Forbes A.P. Antibodies to cytoplasm of ova. Lancet
1966;2:264-5.
46-Irvine W.J., Chan M.M.W, Scarth L. The futher characterization of autoantibodies
reactive with extra-adrenal steroid-producing cells in patients wuth adrenal
disorders. Clin. Exp. Immunol 1969;4:489-503.
47- Scott R.T. and Hofmann G. Prognostic assessment of ovarian reserve.
Fertil.Steril. 1995,63:1-11.
48-Charreau E. Premature ovarian failure and immunity. 7th World Congress
of Gynecol.ogical Endocrinology. 1999, April 25-28, Buenos Aires, Argentina
49- Corenblum B., Rowe T., Taylor P. High-dose, short-term glucocortcoids
for the treatment of infertility resulting from premature ovarian failure.
Fertil. Steril. 1993;59:988-991.
50- Rees Smith B., Furmaniak J. Editorial: Adrenal and gonadal autoimmune
diseases. J.Clin. Endocrinol. Metab. 1995; 80: 1502-1504.
51-Charreau E.H. Conferencia. Antirreceptores en el Síndrome de
ovario resistente. Acta Bioq.Clin.Lat. 1981,51: 251-252.
52-Blussman L., Chiauzzi V. y Charreau E. Anticuerpos anti-ovario y falla
ovárica prematura. Medicina 1998,58:608.
53-Ahonen P., Miettinen A. and Perheentupa J. Adrenal and steroid cell
antibodies in patients with autoimmune polyglandular disease type I and
risk of adrenocortical and ovarian failure. J. Clin. Endocrinol. Metab.
1987, 64:494-500.
AGRADECIMIENTOS:
Este trabajo fue realizado gracias a la col.aboración del Dr. Eduardo
H. Charreau y la Dra. Violeta Chiauzzi.
Instituto de Biología y Medicina Experimental ( IBYME)
Buenos Aires, Argentina.