|
INMUNOGLOBULINAS
Estructura y función: las inmunoglobulinas (Ig) están
constituidas por dos cadenas
pesadas idénticas H y dos cadenas
livianas L, unidas por uniones disulfuro.
Las cadenas H y L tienen los
extremos aminoterminales del mismo lado e incluyen series
de regiones
globulares de gran homología en la secuencia de aminoácidos,
conocidos como dominios.
Los dominios NH2-terminal de las cadenas contienen secuencias
variables de la región V que determina la especifidad antigénica,
en la cual se encuentra el epitope. Además de la región
V, los dominios restantes de las regiones constantes de la cadena H tienen
diferencias estructurales y antigénicas que permiten su clasificación
en subclases.
Heterogeneidad de los anticuerpos: se puede dividir en variaciones
isotípicas, alotípicas e idiotípicas.
Variación isotípica: los isotipos tienen el mismo dominio
constante. Las variaciones isotípicas permiten las diferencias
en clases de H (cadenas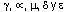 de las IgG, IgA, IgM, IgD y IgE respectivamente), en tipos de L (cadenas
tipo kappa y lambda) y en dominios que están presentes en todos
los miembros sanos de una especie dada. La síntesis de isotipos
está codificada por genes presentes en todos los individuos de
la especie.
de las IgG, IgA, IgM, IgD y IgE respectivamente), en tipos de L (cadenas
tipo kappa y lambda) y en dominios que están presentes en todos
los miembros sanos de una especie dada. La síntesis de isotipos
está codificada por genes presentes en todos los individuos de
la especie.
Variación alotípica: es la variación de una inmunoglobulina
inducida por alelos dentro de una especie. Se presentan como variantes
de la región constante de una cadena H. Por ejemplo, Gm es el factor
responsable de la regulación de una región constante de
la cadena H de IgG. Se conocen 25 marcadores Gm distintos.
Variación idiotípica: los idiotipos tienen el mismo dominio
variable. La variación idiotípica se relaciona con la diversidad
del sitio de unión al antígeno. Las inmunoglobulinas producidas
por un clon celular son completamente idénticas.
Inmunodeficiencias:
Inmunodeficiencias primarias:
Son desórdenes relativamente raros del sistema inmunológico.
Se clasifican de acuerdo a la parte del sistema inmune involucrado y resultan
en una predisposición aumentada a las infecciones.
Los hallazgos de laboratorio más comunes son los siguientes:
Inmunodeficiencia
|
Comentarios |
Déficit de células B
Agammaglobulinemia (tipo Bruton). |
Herencia recesiva ligada al sexo (mutación en el gen BTK).
Recuento linfocitos B CD19 (+) menores de 2%
IgG menor de 1 g/l; otras Ig ausentes. |
Síndrome de deficiencia de anticuerpos variable
no clasificable.
|
Déficit en el sistema de células B
IgG e IgA marcadamente reducidas. IgM muy disminuida.
Los síntomas aparecen recién en la pubertad. |
Síndrome de hiper IgM
|
Hereditario ligado al sexo, mutación en el gen
de la proteína CD40 ligando.
IgG e IgA disminuidas; IgM elevada o normal. No hay switch de IgM
a IgG o IgA en las membranas (deficiencia en la colaboración
de linfocitos T a B). |
Deficiencia selectiva de IgA
|
Reconoce múltiples causas:
-Inhibición de diferenciación de células B.
-Disminución de secreción de IgA por las células
plasmáticas.
-Eliminación inmune por anti-IgA. |
Deficiencia selectiva de IgM
|
Muy rara, asintomática. |
Deleción genética que afecta las Ig.
|
En el cromosoma 14q32. Disminuye una clase de IgG. |
Deficiencia de cadena K
|
Muy rara. |
Déficit de células T
Síndrome de Di George |
El sistema de células T no existe y el B es normal debido a
aplasia de timo o a displasia tímica con aplasia de paratiroides.
Las Ig son normales. |
| Candidiasis mucocutáneas. |
Deficiencia selectiva de células T. Ig normales. |
Síndrome de inmunodeficiencia severa combinada
|
- Herencia autosómica recesiva.
- Herencia ligada al sexo. No hay células B ni T. Ig ausentes
(mutación en el gen de la cadena gama común a los receptores
de IL- 2, 4, 7, 9 y 15. |
Síndrome de Nezelof
|
Herencia autosómica y ligada al sexo. Déficit
de células T por displasia en timo. Ig normales o reducidas. |
Síndrome de Louis-Bar
|
Sistema T afectado. Puede haber deficiencia de IgA
e IgE. Asociada a ataxia telangiectasia. |
Síndrome de Wiskott-Aldrich
|
Herencia ligada al sexo, sólo afecta niños.
(mutación en el gen WASP). IgG normal, IgA e IgE elevados,
IgM disminuida. La forma severa se presenta con trombocitopenia y
eczema. |
En el cuadro siguiente se resume el enfoque diagnóstico para
una inmunodeficiencia primaria:
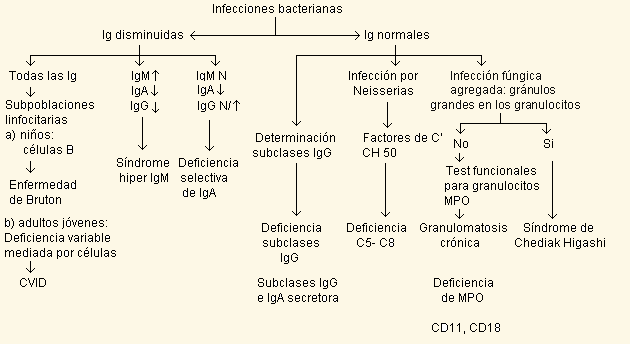
Los criterios clásicos para el diagnóstico se basan en
la clínica, herencia, fenotipo inmunológico y otros estudios.
Actualmente (desde 1998) el diagnóstico se puede hacer a nivel
molecular mediante una técnica de rastreo (SSCP, análisis
de polimorfismo de conformación del ADN de simple cadena) y confirmar
la mutación por secuenciación.
Inmunodeficiencias adquiridas:
Debido a un mejor conocimiento de las interacciones inmunológicas,
se requieren nuevas y más complejas pruebas diagnósticas
de laboratorio que incluyen el estudio de algoritmos en los pacientes
que sufren inmunodeficiencia adquirida.
Para su estudio se define, ante todo, al paciente inmunocomprometido como
aquella persona cuyo sistema inmune revela deficiencias cuantificables
que la causen repetidas enfermedades, es decir, si presenta al menos tres
episodios de infecciones por año de más de 4 semanas de
duración.
Clasificación:
Las inmunodeficiencias adquiridas son enfermedades muy frecuentemente
halladas en práctica clínica, incluyendo tanto las inmunodeficiencias
primarias como las secundarias.
La mayoría de los pacientes con inmunodeficiencia primaria están
afectados desde la infancia y la mayor proporción es de origen
genético. Se conocen alrededor de 100 enfermedades y su característica
principal es la infección recurrente o crónica, producida
por gérmenes oportunistas y de respuesta parcial al tratamiento.
En cambio las inmunodeficiencias adquiridas secundarias, en contraste
con las congénitas, se caracterizan por la presencia de memoria
inmunológica detectable (respuesta débil a IgG o IgA a los
antígenos de las vacunas o las pruebas cutáneas, por ejemplo).
La siguiente es una lista de enfermedades que se presentan con inmunodeficiencias:
Inmunodeficiencias adquiridas:
- A - Primarias, de diagnóstico tardío, descriptas en
el cuadro anterior.
· deficiencia selectiva de IgA
· deficiencia en subclases de IgG
· inmunodeficiencia común variable
· síndrome de hiper-IgM
· enfermedad de Bruton
· síndrome con CD 4 bajo (etiología indeterminada.
Pérdida de CD4+ en infección por HIV)
· déficit de factores de complemento:
C1-C5 (glomerulonefritis)
C5-C9 (sepsis)
C1-inhibidor (edema angioneurótico).
- B - Secundarias, de tipo fisiológico y exógeno
· edad progresiva
· malnutrición
· medio ambiente (carencia de higiene y disbalance dietario,
venenos químicos, radiación UV)
· politraumatismos, quemaduras
- C - Secundarias conjuntamente con otras enfermedades
Infecciones:
virus: EBV, CMV, parvovirus B19, HSV, HCV.
bacterias: micobacterias, Treponema pallidum.
hongos
parásitos (malaria, leishmaniasis, tripanosomiasis, esquistosomiasis).
Enfermedades autoinmunes:
Lupus eritematoso sistémico.
Enfermedad mixta del tejido conectivo.
Glomerulonefritis.
Sarcoidosis
Tumores malignos:
Sistema linfático.
Enfermedad de Hodgkin.
Leucemia linfocítica aguda y linfocítica crónica.
Mieloma.
Tumores sólidos.
Síndromes metabólicos:
Diabetes mellitus.
Enfermedad de Cushing.
Uremia.
Síndrome de depleción proteica.
Linfangiectasia intestinal.
Síndrome de Down.
Iatrogénica:
Esplenectomía
Cirugías extensivas.
Terapia citotóxica e inmunosupresora.
Transplante de médula ósea.
Enfoque diagnóstico: el diagnóstico depende casi
totalmente del laboratorio. Los pasos a seguir para una evaluación
diagnóstica de un paciente con inmunodeficiencia son los siguientes:
1 - Anamnesis.
2 – Tests de laboratorio:
- Eritrosedimentación, leucocitos, funcionalismo renal, proteínas,
glucosa.
- Cuantificación de IgM, IgG, IgA, IgE.
- Ig específicas: HIV, HBV, HCV, EBV, CMV.
- Sistema de complemento.
- Proteína C reactiva.
- Autoanticuerpos (ANA, ANCA, F.R.).
- Tests cutáneos estandarizados (Multitest - Merieux).
- Aislamiento en cultivo de agentes patógenos.
3.- Estado inmune completo:
· Fenotipificación de células T, B y NK y de
monocitos y granulocitos.
· Respuesta linfoproliferativa a antígenos, mitógenos,
producción de citoquinas, función linfocitotóxica.
· Presentación de antígeno por células presentadoras.
· Síntesis in vitro de IgG por mitógenos de células
B dependientes e independientes de células T.
Bibliografía
1. LotharT. Clinical Laboratory Diagnostics: Use and assessment of clinical
laboratory results, English edition, 1998.
2. Jacobs D.S., Demott W.R. Grady H. et al., Laboratory Test Handbook,
Edit by Lexi-Comp Inc., Cleveland, United States of America, 4th edition,
1996.
3. Tietz N. W. Clinical Guide to Laboratory test, edited by W.B. Saunders
Company, third edition, United States of America ,1995.
4. Celasco M., Rossi J., Danielia S. “El laboratorio inmunológico:
Aportes al diagnóstico y seguimiento de inmunodeficiencias primarias
genéticas y adquiridas”. 1er Congreso ALAC. 69ª Reunión.
25 al 28 de abril 2001.
5. Young D. Effects of Preanalytical Variables on Clinical Laboratory
Test . AACC, second edition, 1997.
6. Young D. and Friedman R. Effects of Disease on Clinical Laboratory
Test, edited by AACC, third edition, 1997.
7. Lehmann C. A. Saunders Manual of Clinical Laboratory Science, W.B.Saunders
Company, Philadelphia, first edition 1998.
8. Young D. Effects of Drugs on Clinical Laboratory Test, AACC, third
edition, 1990.
9. Lockitch G. Handbook of Diagnostic Biochemistry and Hematology in Normal
Pregnancy. CRC Press, Inc. United States of America, 1993
|